





所有平台仅提供服务对接功能,所载文章、数据仅供参考,股市有风险,投资需谨慎,用户需独立做出投资决策,风险自担!

时间:2022-09-07 14:03:52来源:网络整理
1.本发明属于纳米生物材料领域,具体涉及一种透明质酸衍生物包覆磷酸钙纳米粒子的制备方法及应用。
背景技术:
2.磷酸钙纳米粒子作为一种具有良好生物安全性和可降解性的天然材料,在转染核酸药物(siRNA、DNA、mRNA等)治疗疾病的研究中表现出良好的递送效率然而,磷酸钙纳米颗粒在生理环境中快速且不可控的生长,使其难以在肿瘤等病灶中蓄积,严重限制了其临床应用。
3.针对磷酸钙纳米粒粒径增长不可控的问题,目前改进的主要方法是以磷酸钙/核酸药物纳米沉淀为核心,粒径不断增长受到连接键覆盖的阻碍 保护性外层主要包括脂质材料(lipid)、peg、粘多糖和plga等无机材料。比较具有代表性的是利用脂质层控制磷酸钙纳米粒的粒径增长,利用反相微乳液法制备核酸药物/磷酸钙纳米粒(脂质包覆磷酸钙-1,lcp-1)其中,反相微乳液是将水溶液分散到含有壬基酚聚氧乙烯醚(igepal-co-520)的环己烷油相溶液中,主要分为三个步骤:1)分散cacl2溶液(ph=9)和核酸药物/nahpo4(ph=9)混合溶液分别进入油相,将两相混合搅拌,微乳液交换反应产生siRNA包裹的钙磷酸钙沉淀;2)加入柠檬酸钠至溶液澄清,稳定磷酸钙纳米粒子(粒径约80nm),同时使纳米粒子表面带负电荷,有利于结合与阳离子脂质体结合。乙醇和水洗脱 硅胶柱法纯化; 3)进一步与阳离子磷脂1,2-二油氧基3-三甲基氨基丙烷(dotap)/胆固醇反应,得到粒径约150 nm的lcp-1纳米粒子。
4.但是,上述制备方法存在两大缺陷:1)操作过程复杂; 2)有机溶剂的使用成本很高。
技术实施要素:
5.发明目的:本发明对现有技术存在的问题进行改进,即公开了一种透明质酸衍生物包覆磷酸钙纳米粒子的制备方法及应用。本发明首先合成透明质酸衍生物材料,然后通过简单的混合方法构建包覆透明质酸衍生物的磷酸钙纳米粒子。更便宜、更安全。
6.技术方案:透明质酸衍生物包覆磷酸钙纳米粒子的制备方法,包括以下步骤:(1)透明质酸衍生物的合成:(11)添加0.3~0.7摩尔份的透明质酸和0.9~1.8摩尔份的羧基活化剂加入反应釜,然后用溶剂完全溶解后,混合得到溶液,将混合溶液的pH值调至4~5;(12)向反应器中加入0.3至0.7摩尔份的双膦酸盐小分子化合物,然后在18-25℃反应24-72 h,取透析后的滤液,用冷冻干燥机冷冻干燥滤液,得到透明质酸衍生物;(2)透明质酸衍生物的制备包覆磷酸钙纳米粒子:
(21)将1份体积的cacl2水溶液和1份体积的核酸药物水溶液混合成溶液;(22)混合2份体积的缓冲液和2体积份将步骤(1)得到的透明质酸衍生物水溶液等体积混合均匀,得到b液;(23)步骤(21)@)得到的a液>滴加到步骤(2<在@k9@得到的溶液b中37-40℃孵育0.5-1 h后得到透明质酸衍生物包覆的磷酸钙纳米粒子。
7.进一步,步骤(11)所述的透明质酸的分子量为48kd-780kd,优选48kd。
8.进一步,步骤(11)中的羧基活化剂为hbtu与1-羟基苯并三唑等摩尔比的混合物,或:等摩尔比为1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐和n-羟基琥珀酰亚胺。
9.进一步,步骤(11)所述的溶剂为去离子水、dmf、dmso中的一种,优选去离子水。
10.进一步,在步骤(11)中,将混合物的pH值调整为4.8。
11.进一步,步骤(12)所述的双膦酸盐小分子化合物为阿仑膦酸钠和帕米膦酸二钠中的一种,优选阿仑膦酸钠。
12.进一步,步骤(21)所述的cacl2水溶液的浓度为100-500mmol
•
l-1
,优选250mmol
•
l-1
.
13.进一步,步骤(21)所述的核酸药物水溶液中的溶质为siRNA、mrna、dna中的一种。
14.进一步,步骤(21)中核酸药物水溶液的浓度为1-50μmol
•
l-1
.
15.进一步,步骤(22)中所述的缓冲区为hepes缓冲区或pbs缓冲区,优选为hepes缓冲区。
16.进一步地,步骤(22)的透明质酸衍生物水溶液的浓度为0.5~1.5mg/ml。
17.进一步地,步骤(23)中包覆有透明质酸衍生物的磷酸钙纳米粒子中的磷酸钙包括磷酸三钙、磷酸氢二钙和磷酸二钙。氢钙和磷酸八钙。
18.上述方法制备的透明质酸衍生物包覆磷酸钙纳米粒在制备治疗肿瘤药物中的应用。
19.上述方法制备的透明质酸衍生物包覆磷酸钙纳米粒在制备治疗关节炎药物中的应用。
20.有益效果:本发明公开的透明质酸衍生物包覆磷酸钙纳米粒的制备方法和应用的优点是:1、核酸药物磷酸钙核外包覆透明质酸酸衍生物,抑制核心的不可控生长,粒径均匀稳定在200nm左右; 2、混合法制备,时间短,成本低,无需实用有机溶剂,清洁无污染;3、生物相容性较好,所用材料为临床常用药物透明质酸辅料,阿仑膦酸钠,临床用药,安全性好。
图纸说明
21.图1是透明质酸-阿仑膦酸盐(hl)的1H NMR图谱。
22.图2为不同阿仑膦酸钠修饰的hl@cap/siRNA的粒径分布。
23.图3是hl@cap/siRNA纳米粒子的dls粒径图(p%=80%)。
24.图4是阿仑膦酸钠修饰的hl@cap/siRNA的透射电镜图。
25.图5为阿仑膦酸钠修饰的hl@cap/siRNA对siRNA的血清保护作用。
26.图6是阿仑膦酸钠修饰的hl@cap/siRNA的放置稳定性示意图。
27.图7为阿仑膦酸钠修饰的hl@cap/siRNA的稀释稳定性图。
28.图8是hl@cap/siRNA与市售转染试剂lipofectamine2000细胞摄取能力比较示意图。
29.具体实施方式:下面对本发明的具体实施方式进行详细说明。
30.实施例1:透明质酸衍生物包覆磷酸钙纳米粒子的制备方法,包括以下步骤:(1),合成透明质酸-阿仑膦酸钠(hl)首先加入200 mg(0.5 mmol)透明质酸(ha)(分子量48 kda)于100 ml茄形烧瓶中,加入20 ml去离子水溶解,放入edc 143.85 mg (0.75 mmol)和NHS 86.25 mg (0.75 mmol),完全溶解后,调节pH至4-5活化0.5 h;后即称取阿仑膦酸钠162.56 mg(0.5 mmol)加入上述混合物,室温反应24 h(pH维持在4-5),透析除去未反应的阿仑膦酸钠和杂质等,用冷冻干燥机冷冻干燥得到样品,得到的样品用核磁共振技术(1h nmr)表征,得到氢核磁共振谱(图 1). <@k 4@>。从图1可以看出,ha上的h分布为1.8~2.0 ppm(甲基,c峰)和3.0~4.0ppm(其他)。通过将阿仑膦酸盐与 HA 连接合成的 Aha 出现了新的分布:~1.7 ppm(亚甲基,靠近糖环,峰 b)和~2.7 ppm(两个亚甲基,靠近磷酸基团,峰),表明阿仑膦酸盐与透明质酸成功连接。经过上述试验分析,目标产物得到确认。
31.(2),hl@cap/sirna纳米粒子的制备首先,10 μl cacl2 (250 mmol
•
l-1
) 和 10 μl siRNA (4.8 μmol
•
l-1

) 等体积混合均匀(ph7.27) 得到溶液;然后加入 20 μl hepes 缓冲液(hbs, 50 mmol
•
l-1 hepes, 280 毫摩尔
•
l-1 nacl,1.5 毫摩尔
•
将不同浓度的
l-1 na2hpo4、ph7.27)和20 μl hl等体积混合,得到溶液b;将a液滴加到b液中,37℃孵育1 h,得到hl@cap/siRNA纳米颗粒。
32.hl@cap/sirna纳米粒子(1)的表征,使用马尔文粒度仪(dls)测量粒度分布和电位,得到图2。图2,siRNA和磷酸钙沉淀凝固形成核,然后hl通过核表面的阿仑膦酸钠与钙离子的相互作用吸附在核周围,形成核壳结构,阻碍了核的进一步生长。核心。
33.(2),为了研究hl添加对磷酸钙纳米粒子生长的影响,测定了不同hl引入量下hl@cap/sirna纳米粒子的粒径随着hl引入量的增加,粒径逐渐减小,当p%大于等于70%时,粒径可以控制在200 nm左右,说明hl可以有效抑制不规则磷酸钙纳米粒子的生长. 基于此, hl 的选择 对@cap/siRNA 纳米粒子 (p%=80%) 进行后续实验, 如图 3 所示, 粒径约为 195 nm 和电位约为-15 mv,而未修饰的磷酸钙纳米粒子(cap/siRNA)粒径约为3200 nm,电位接近中性。
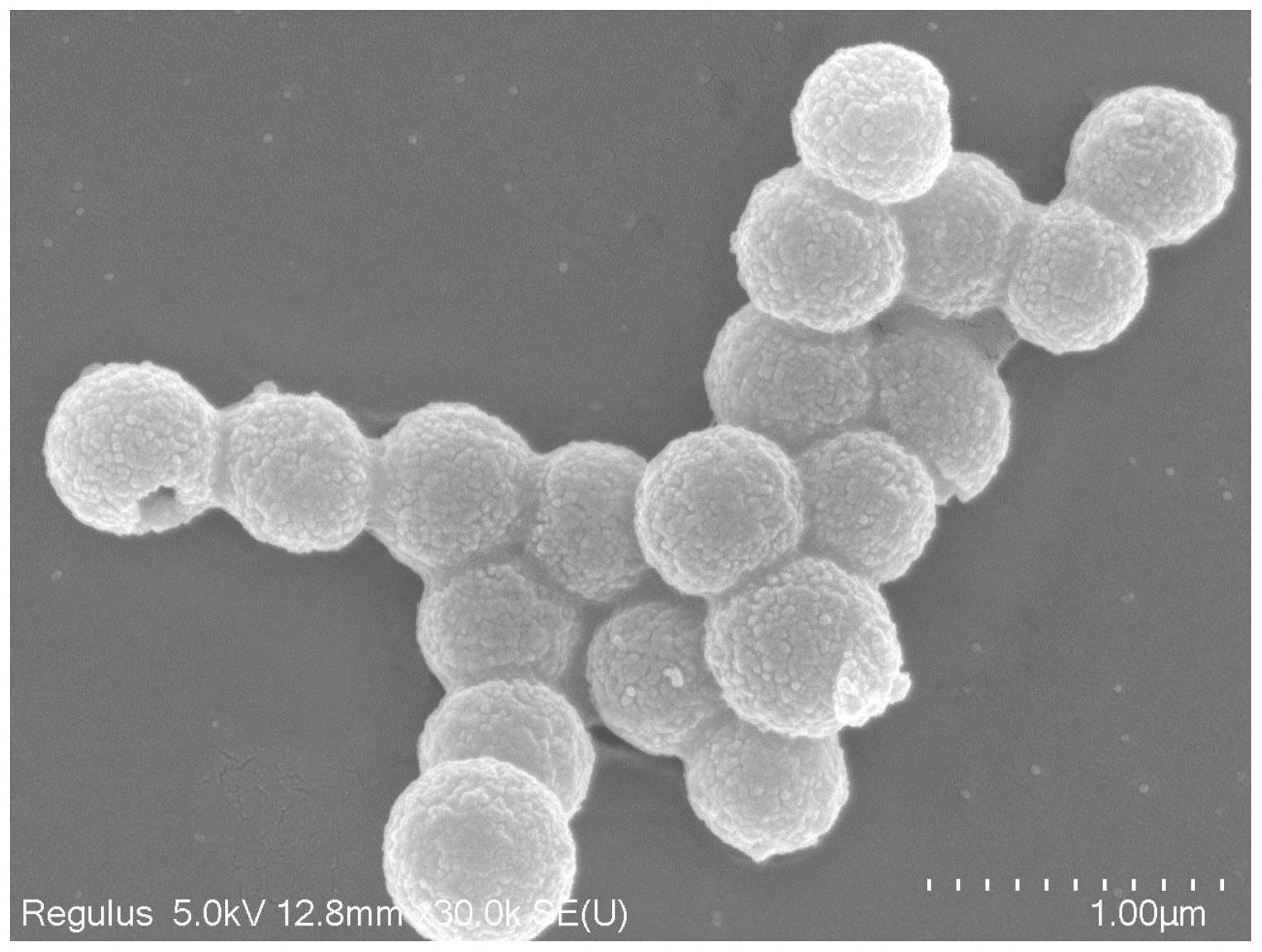
34.(3),取10 μl经过筛网优化的hl@cap/sirna纳米颗粒,滴在碳包铜网上,自然蒸发晾干,使用joel 100cx透射电镜(电压100kv)观察拍照,得到图4。如图4所示,
cap/siRNA 纳米粒子聚集成团块,粒径大于 1 μm,而 hl@cap/siRNA 纳米粒子 (p%=80%) 大多单独分散,呈球形,大小约 150 nm,略小于水合颗粒的大小是因为电子显微镜观察到水挥发后收缩的颗粒。
35.(4), hl@cap/sirna 纳米粒子的稳定性 检查血清中 hl@cap/sirna 纳米粒子的稳定性:保护 siRNA 免受 RNAase 降解 游离 siRNA, cap/sirna nps 和 hl@cap/sirna nps 与等体积的胎牛血清混合(最终 siRNA 浓度为 1.2 μm),在 37°C 下孵育 0 h、1 h、2 h、4 h 、6小时、9小时、12小时、24小时、36小时、48小时采样,-20度保存。
36.取完所有样本后,每个时间点的样本加6
含1mol/l hcl
×
RNA 上样缓冲液,在含有 0.5 μg/ml 凝胶红的 1% 琼脂糖凝胶上在 80 v 下电泳 3 分钟,然后在 100 v 下电泳 15 分钟,imagerquant rt ecl 凝集 Gel red/siRNA使用凝胶成像系统观察荧光。内环境和细胞质中含有较多的RNA酶,可以破坏siRNA,使其失去基因沉默作用。大多数运载载体在包裹siRNA后可以显着增强siRNA对RNase降解的抵抗力。结果如图5所示。通过凝胶电泳实验研究了不同纳米粒子对siRNA的保护作用。游离的siRNA在2小时内基本被RNAse完全降解,cap/siRNA纳米颗粒可以有效保护siRNA约24小时,hl@cap/siRNA纳米颗粒对siRNA的保护作用更强,并且仍然保持在48 小时。可以观察到清晰的条带。 hl@cap/sirna纳米粒子对siRNA更好的保护作用也表明它们可以有效地包裹siRNA。
37.hl@cap/sirna纳米粒子在室温下的稳定性研究1、3、5、7、10、@ >30天,测量粒径磷酸钙沉淀,如图6所示,hl@cap/siRNA纳米粒子的粒径随时间保持稳定。
38.hl@cap/sirna纳米粒子稀释稳定性考察:取20 μl hl@cap/sirna纳米粒子,用0.9%生理盐水稀释至原体积8、16、32、64、128、256 和 512 次,使用 Malvern 激光粒度仪测量粒度变化。如图7所示,随着稀释倍数的增加,hl@cap/siRNA纳米粒子的粒径变化不大,表明它们具有更好的抗稀释能力。
39.(5),人非小细胞肺癌细胞a549和人非小细胞肺癌细胞a549对hl@cap/siRNA纳米粒子的摄取能力为250,000每孔,分别在六孔板中,24小时后更换培养基,加入opti-mem培养基,分别用不同的纳米复合物转染fam-siRNA,消化并离心,PBS洗涤两次,300
ꢀµ
l PBS重悬,立即用流式细胞仪测定细胞内fam的荧光强度。收集了 10,000 个细胞。激发波长为 488 nm,发射波长为 518 nm。使用fcs expressv3软件对采集的数据进行分析,得到图8。 如图 8 所示,hl@cap/siRNA 纳米粒的摄取是未修饰组(cap/siRNA 纳米粒)的 3 倍,说明 hl 修饰可以显着促进细胞对 siRNA 的摄取,可用于后续高效基因沉默。为更好的抗肿瘤潜力奠定了基础。
40.实施例2 透明质酸衍生物包覆磷酸钙纳米粒子的制备方法,包括以下步骤:(1)透明质酸衍生物的合成:(11)添加<将@0.3摩尔份的透明质酸和0.9摩尔份的羧基活化剂加入反应器,然后用溶剂完全溶解得到混合溶液,然后调节混合溶液的pH值(可用氢氧化钠水溶液调节pH值)至4;(12)向反应器中加入0.3摩尔份的双膦酸盐小分子化合物,然后在18℃进行反应取出272 h,透析后收集滤液,用冷冻干燥机冷冻干燥,得到透明质酸衍生物;
(2)透明质酸衍生物包覆磷酸钙纳米粒子的制备:(21)将1体积份的cacl2水溶液和1体积份的核酸药物水溶液混合,得到溶液; (22)将缓冲液2体积份和步骤(1)得到的透明质酸衍生物水溶液2体积份混合,得到b溶液;(23)将步骤(21)得到的溶液a滴加到步骤(22)得到的溶液b中,然后37℃孵育1h,得到包覆有透明质酸衍生物的磷酸钙纳米粒子。
41.进一步,步骤(11)的玻尿酸分子量为48kd。
42.进一步,步骤(11)中所述的羧基活化剂为等摩尔比的hbtu (o-benzotriazole-n,n,n',n'-Tetramethylurea hexafluorophosphate,cas number : 94790-37-1)和1-羟基苯并三唑的混合物。
43.进一步磷酸钙沉淀,步骤(11)所述的溶剂为去离子水。
44.进一步,步骤(12)所述的双膦酸盐小分子化合物为阿仑膦酸钠。
45.进一步,步骤(21)所述的cacl2水溶液的浓度为100mmol
•
l-1
.
46.进一步,步骤(21)所述的核酸药物水溶液中的溶质为siRNA。
47.进一步,步骤(21)为1μmol的核酸药物水溶液浓度为1μmol
•
l-1
.
48.进一步,步骤(22)所述的缓冲液为hepes buffer hepes buffer (50mmol
•
l-1
hepes,280 毫摩尔
•
l-1
nacl,1.5mmol
•
l-1
na2hpo4, ph7.27).
49.进一步,步骤(22)的透明质酸衍生物水溶液的浓度为0.5mg/ml。

50.进一步地,所述步骤(23)包覆透明质酸衍生物的磷酸钙纳米粒子中的磷酸钙包括磷酸三钙、磷酸氢二钙、磷酸二钙、氢钙和磷酸八钙) .
51.上述方法制备的透明质酸衍生物包覆磷酸钙纳米粒在制备治疗肿瘤药物中的应用。
52.上述方法制备的透明质酸衍生物包覆磷酸钙纳米粒在制备治疗关节炎药物中的应用。
53.实施例3 透明质酸衍生物包覆磷酸钙纳米粒子的制备方法,包括以下步骤:(1)透明质酸衍生物的合成:(11)添加<将@0.7摩尔份的透明质酸和1.8摩尔份的羧基活化剂加入反应器,然后用溶剂完全溶解得到混合溶液,然后调节混合溶液的pH值(pH值可用氢氧化钠水溶液调节)至5;(12)向反应器中加入0.7摩尔份的双膦酸盐小分子化合物,然后在25℃进行反应取出24小时,透析后收集滤液,将滤液用冷冻干燥机冷冻干燥,得到透明质酸衍生物;(2)透明质酸衍生物包覆磷酸钙纳米粒子的制备:(21)@ > 将1体积份的cacl2水溶液和1体积份的核酸药物水溶液混合n 获得解决方案; (22)2体积份的缓冲液和2体积份的步骤(1)得到的透明质酸衍生物水溶液等体积混合,得到b液;(23)加将步骤(21)得到的a液滴入步骤(22))得到的b液中,40℃孵育0.5h,得到包覆有透明质酸衍生物。
54.进一步,步骤(11)的玻尿酸分子量为780kd。
55.进一步,步骤(11)中所述的羧基活化剂为等摩尔比的1-(3-二甲基氨基丙基)-3-乙基
碳二亚胺盐酸盐和n-羟基琥珀酰亚胺的混合物。
56.进一步,步骤(11)所述的溶剂为dmf。
57.进一步,步骤(12)所述的双膦酸盐小分子化合物为帕米膦酸二钠。
58.进一步,步骤(21)所述的cacl2水溶液的浓度为500mmol
•
l-1
.
59.进一步,步骤(21)所述的核酸药物水溶液中的溶质为mRNA。
60.另外,步骤(21)中核酸药物水溶液的浓度为50μmol
•
l-1
.
61.进一步,步骤(22)中描述的缓冲区为pbs缓冲区。
62.进一步,步骤(22)的透明质酸衍生物水溶液的浓度为1.5mg/ml。
63.进一步地,步骤(23)中包覆有透明质酸衍生物的磷酸钙纳米粒子中的磷酸钙包括磷酸三钙、磷酸氢二钙和磷酸二钙。氢钙和磷酸八钙。
64.上述方法制备的透明质酸衍生物包覆磷酸钙纳米粒在制备治疗肿瘤药物中的应用。
65.上述方法制备的透明质酸衍生物包覆磷酸钙纳米粒在制备治疗关节炎药物中的应用。
66.实施例4 透明质酸衍生物包覆磷酸钙纳米粒子的制备方法,包括以下步骤:(1)透明质酸衍生物的合成:(11)添加<将@0.5摩尔份的透明质酸和1.5摩尔份的羧基活化剂加入反应器,然后用溶剂完全溶解得到混合溶液,然后调节混合溶液的pH值(可用氢氧化钠水溶液调节PH值)至4.8;(12)向反应器中加入0.5摩尔份的双膦酸盐小分子化合物,然后反应22℃48小时,透析后取滤液,用冷冻干燥机冷冻,得到透明质酸衍生物;(2)透明质酸衍生物包覆磷酸钙纳米粒子的制备:(21) @>将1份体积的cacl2水溶液和1份体积的核酸药物水溶液混合,得到溶液;(2 2)将2体积份的缓冲液和2体积份的步骤(1)得到的透明质酸衍生物水溶液等体积混合,得到b液; (23)将步骤(21)得到的液体a滴加到步骤(22))38℃孵育后得到透明质酸衍生物包覆的磷酸钙纳米粒子0.75 小时。

67.进一步,步骤(11)的玻尿酸分子量为350kd。
68.进一步,步骤(11)中所述的羧基活化剂为等摩尔比的hbtu(邻苯并三唑-n,n,n',n'-四甲基脲六氟磷酸酯,cas号: 94790-37-1)和1-羟基苯并三唑的混合物。
69.进一步,步骤(11)中所述的溶剂为脱DMSO。
70.进一步,步骤(12)所述的双膦酸盐小分子化合物为阿仑膦酸钠。
71.进一步,步骤(21)所述的cacl2水溶液的浓度为250mmol
•
l-1
.
72.进一步,步骤(21)所述的核酸药物水溶液中的溶质为dna。
73.另外,步骤(21)中核酸药物水溶液的浓度为25μmol
•
l-1
.
74.进一步,步骤(22)所述的缓冲液为hepes缓冲液(50 mmol
•
l-1 hepes, 280 毫摩尔
•
l-1 nacl,1.5 毫摩尔
•
l-1 na2hpo4, ph 7.27).
75.进一步,步骤(22)中透明质酸衍生物水溶液的浓度为1mg/ml。
76.进一步地,步骤(23)中包覆有透明质酸衍生物的磷酸钙纳米粒子中的磷酸钙包括磷酸三钙、磷酸氢二钙、磷酸二钙、氢钙和磷酸八钙。 .
77.上述方法制备的透明质酸衍生物包覆磷酸钙纳米粒在制备治疗肿瘤药物中的应用。
78.上述方法制备的透明质酸衍生物包覆磷酸钙纳米粒在制备治疗关节炎药物中的应用。
79.综上所述,从本发明实施例可以看出,本发明合成了一种透明质酸-阿仑膦酸钠共轭高分子材料(hl)分步混合法。包覆在磷酸钙纳米沉淀物周围,构建包覆透明质酸衍生物的新型磷酸钙纳米粒子(hl@cap/sirna nps),纳米粒子均匀稳定,操作更简单,不涉及有机溶剂,成本低,为解决siRNA磷酸钙纳米载体突破性应用困境提供了新的策略和新技术。
80.上面对本发明的实施例进行了详细描述。然而,本发明不限于上述实施例,在本领域技术人员所拥有的知识范围内可以进行各种改变而不脱离本发明的精神。
声明:文章仅代表原作者观点,不代表本站立场;如有侵权、违规,可直接反馈本站,我们将会作修改或删除处理。







图文推荐

2022-09-07 13:06:13

2022-09-07 13:01:11

2022-09-07 12:23:14

2022-09-07 11:40:12

2022-09-07 11:03:03

2022-09-06 19:33:03
热点排行
精彩文章

2022-09-07 13:06:06

2022-09-07 12:23:05

2022-09-07 12:02:32

2022-09-07 12:01:51

2022-09-07 11:40:16

2022-09-07 10:57:18
热门推荐
